TeGnostiX helps scientists transform massive biological datasets into meaningful insights by leveraging advancements in next-generation sequencing. We develop automated analysis pipelines and provide services across three key areas: classical data analysis, machine learning for patient stratification and biomarker discovery, and bioinformatics app development. Our classical data analysis includes genomics, transcriptomics, epigenomics, and other omics data, while our machine learning solutions focus on predicting disease pathogenicity. Additionally, we create web and mobile applications for analyzing and visualizing biological data, supporting researchers in deriving impactful conclusions from complex data.
Make use of the revolution in the technology and:
Integrate multi-omics data to identify molecular interactions and disease mechanisms.
Develop predictive models for human pathology and disease classification using omics data.
Characterize and predict cell types, including specific tumor and infection profiles.
Predict biomarkers for diseases, specific cell types, or tumor subtypes.
Identify druggable targets within gene pathways for therapeutic development.
Conduct tissue, cell type, and disease-specific infection analysis.
Compare novel research findings against existing published data for enhanced insights.
Develop web and mobile apps for visualizing biological data and conducting research surveys.
Service
We provide diverse nature of services from classical data analysis to the development of deep learning models for biomarkers identification. We also develop apps for biological data. The Key Features of TeGnostiX
CLINICAL DATA ANALYSIS
CLINICAL DATA ANALYSIS
- Identification of influential factors.
- Impact of age and sex on disease state.
- Meta-analysis of clinical measurements and disease association.
GENOMICS DATA ANALYSIS
GENOMICS DATA ANALYSIS
- Variant calling (SNPs, indels).
- Copy number analysis (gene copy numbers).
- Genomic rearrangements.
- Unkown genome assembly.
TRANSCRIPTOMICS DATA ANALYSIS
TRANSCRIPTOMICS DATA ANALYSIS
- Single cell, Bulk and Small RNA (sRNA) sequencing analysis.
- Cell type annotation and trajectory analysis.
- Differential expresson analysis.
- Pathway analysis.
EPIGENOMICS DATA ANALYSIS
EPIGENOMICS DATA ANALYSIS
- DNA methylation data analysis.
- Histone modification data analysis.
PROTEOMICS DATA ANALYSIS
PROTEOMICS DATA ANALYSIS
- Mass spectrometry data analysis.
- Enrichment & pathway analysis.
- Protein protein-interactions.
- Protein domain and motif analysis
MICROBIOME DATA ANALYSIS
MICROBIOME DATA ANALYSIS
- Taxonomic and functional annotation.
- Metagenome assembly.
- Diversity analyses.
BIOMARKER PREDICTION
BIOMARKER PREDICTION
- Identification of risk and disease biomarkers based on transcripomics, proteomics, GWAS, epigenomics and microbiome.
DEEP LEARNING MODELS
DEEP LEARNING MODELS
- Pathogenicity prediction.
- Patients stratification based on clinical and molecular signatures.
- Cell type prediction.
- Marker genes prediction.
BIOINFORMATICS APP DEVELOPMENT
BIOINFORMATICS APP DEVELOPMENT
- Website development for biological data.
- Database development for biological data.
- Comparison with publicly available data.
- Genome browser development for biological data.
- Mobile app development for biological data.
FEATURED PROJECTS
Selected research highlights.
-
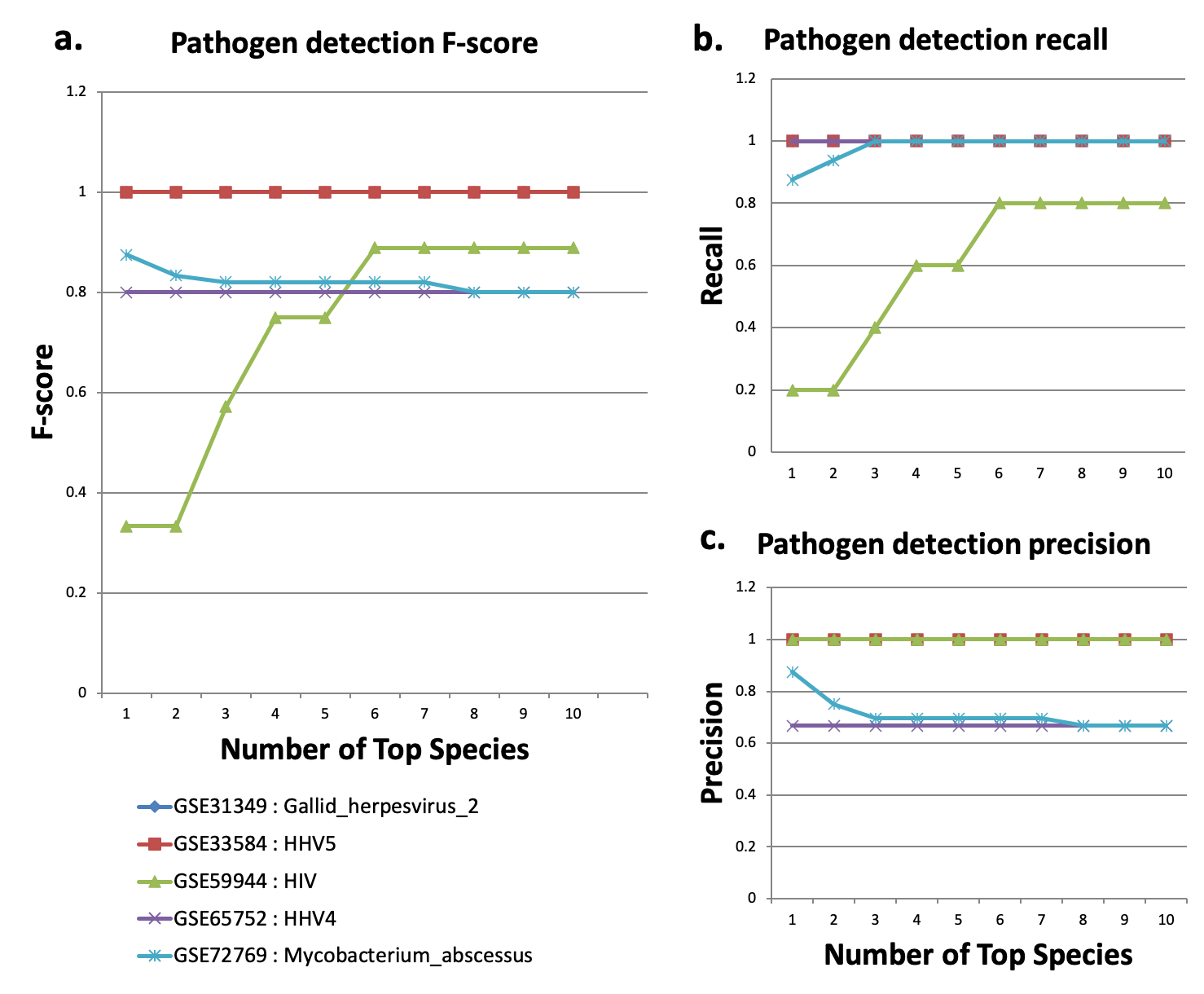
Pathogen detection.
Pathogen detection in sequencing data using Oasis2.
-
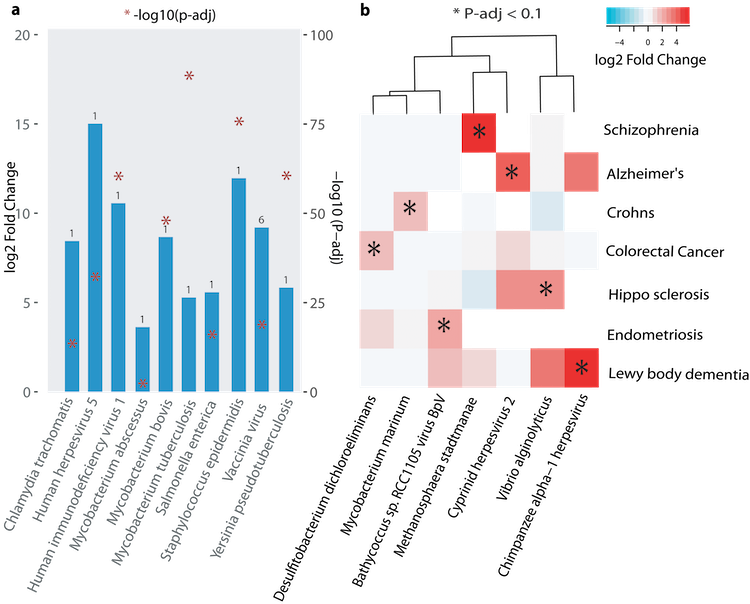
Bacterial and viral infections
Pathogenic biomarker signatures.
-
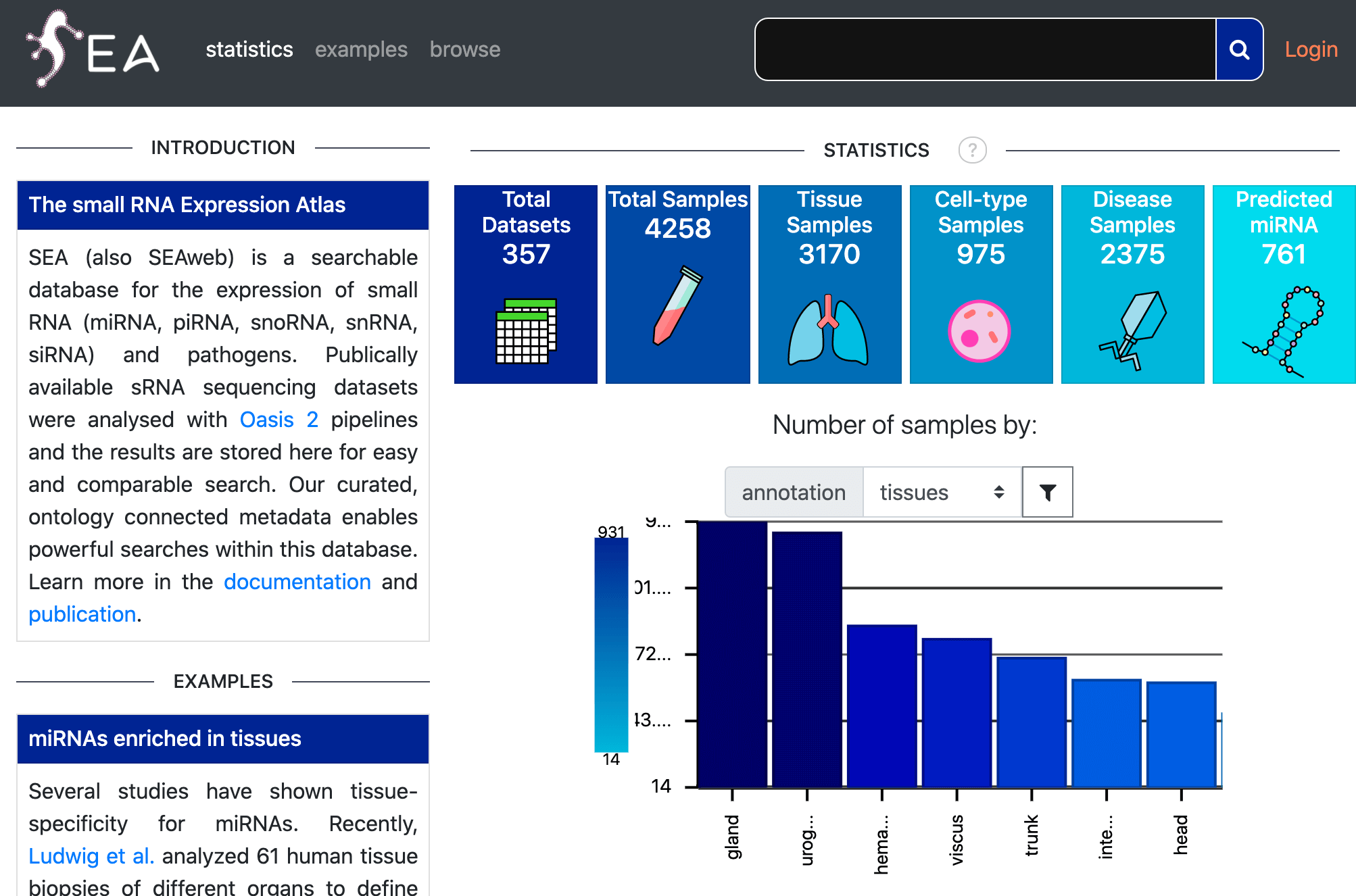
SEA
The Small RNA Expression Atlas.
-
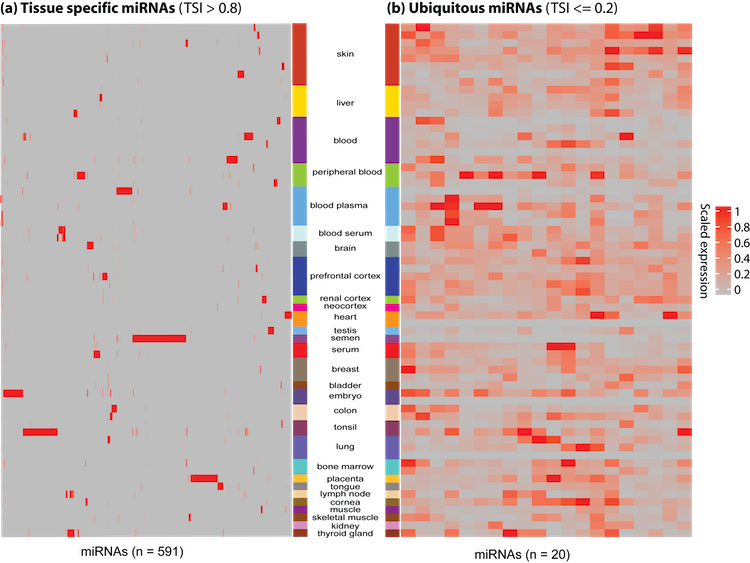
micro RNA tissue specificity
Traditional biomarkers lack tissue specificity.
-
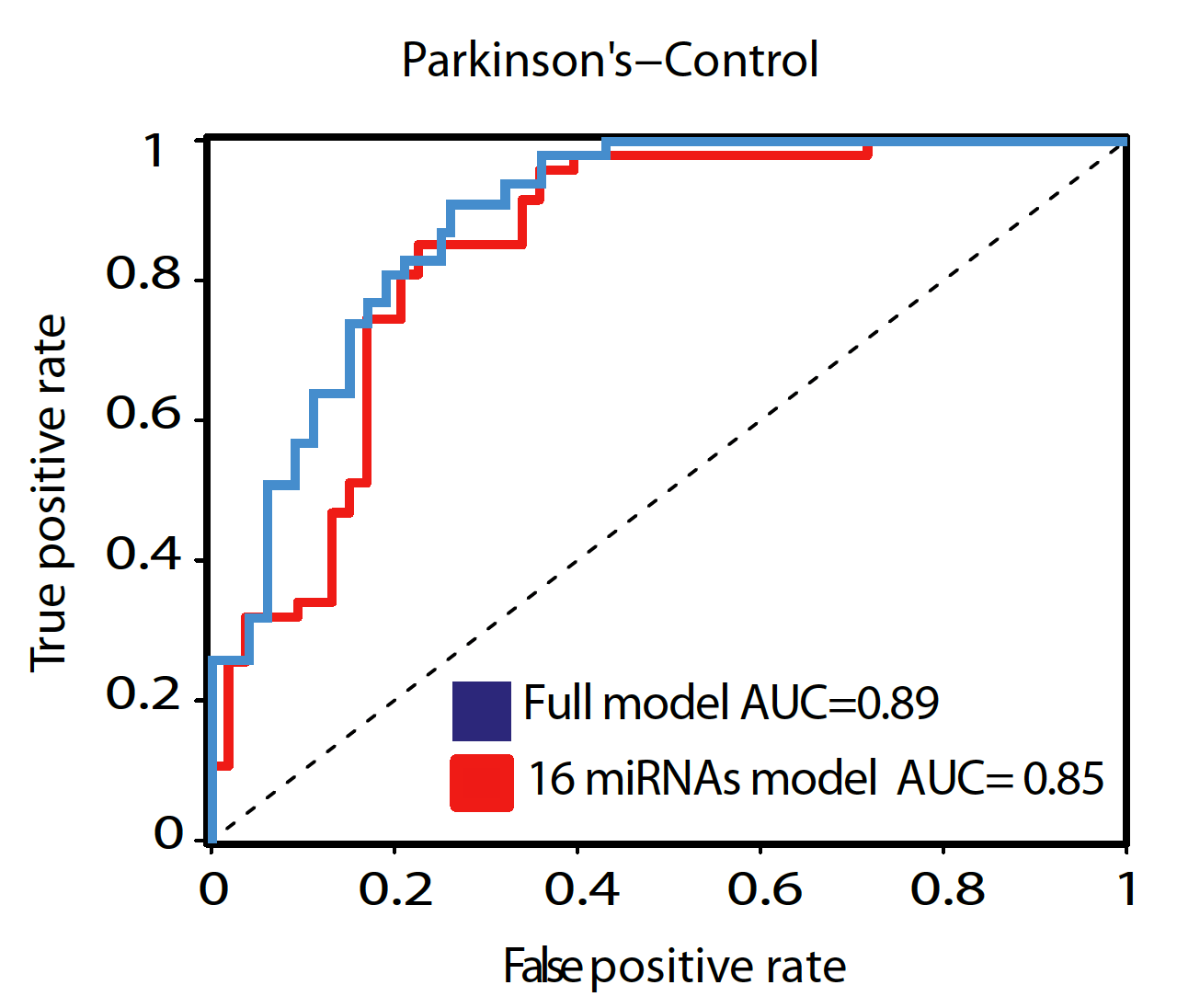
Parkinson disease biomarkers
-
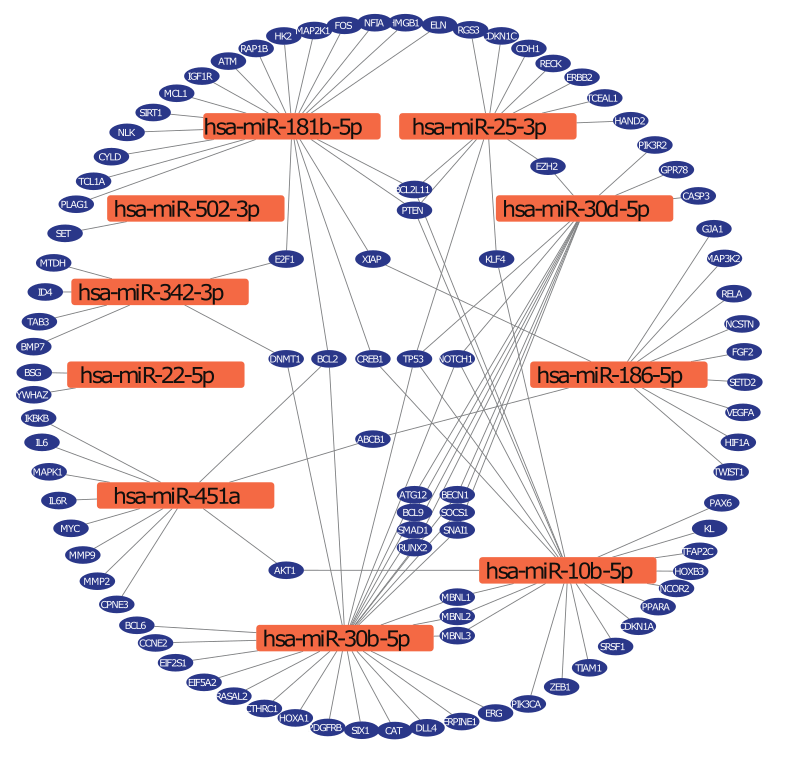
Interaction network
Parkinson's disease associated miRNAs and target genes.
-
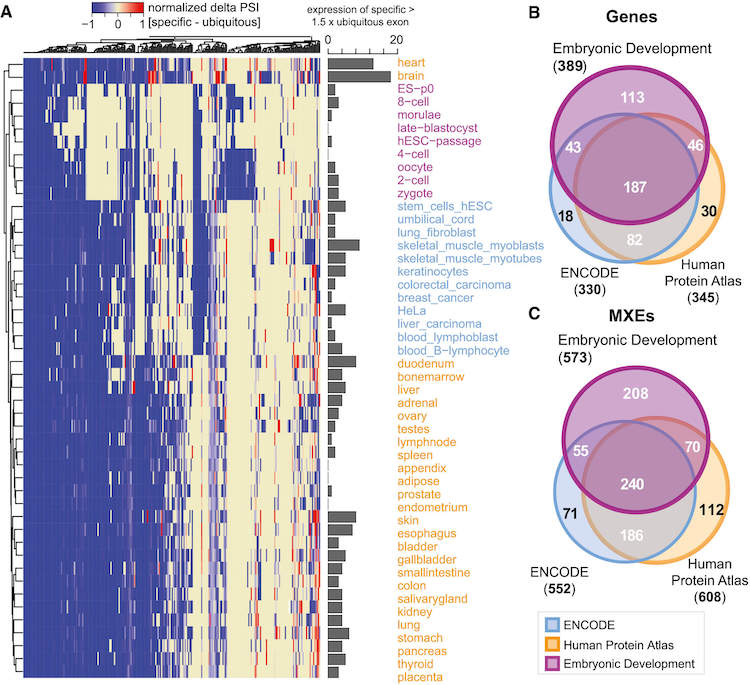
MXEs.
MXEs are tissues & developmental stages specific.
-
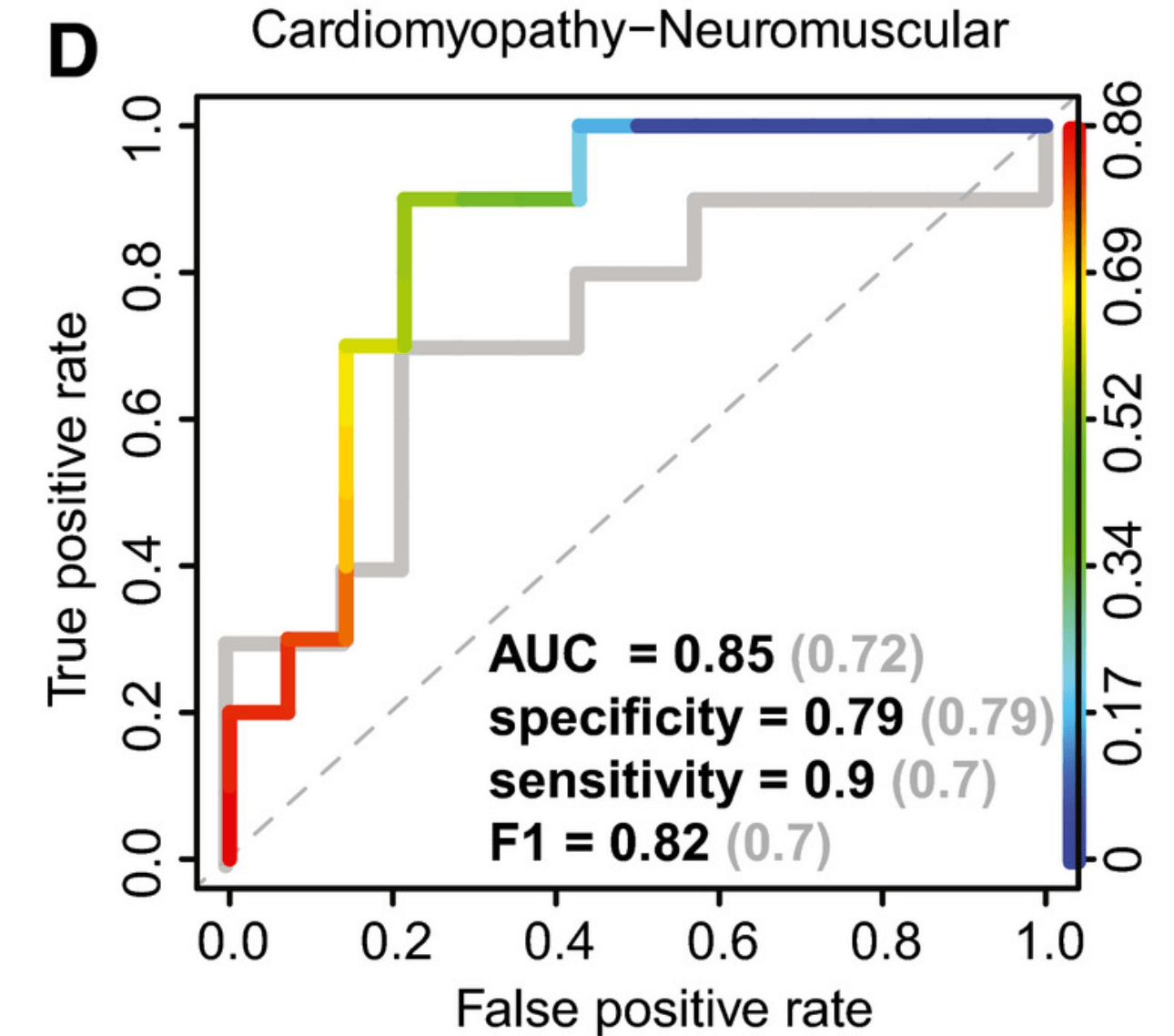
MXE pathogenicity
Can MXE expression predict pathogenicity?
-
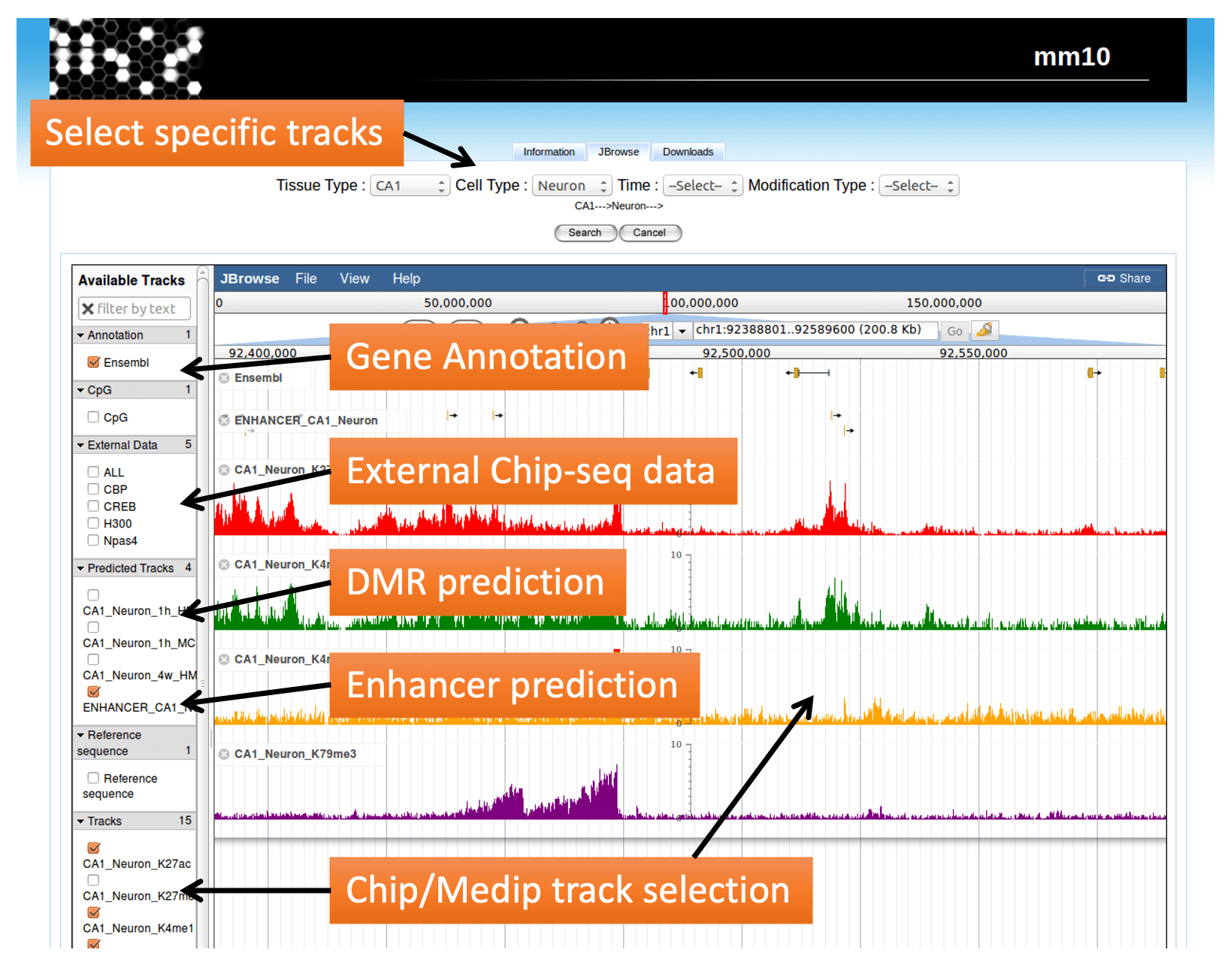
Genome Browser
Make it easy to visualize.
Contact
Leave a Message
Contact Us
TeGnostiX388 Bridle Path, Phone: +1-617-2299738 info (at) TeGnostiX (dot) com
Worcester, MA, 01604
USA